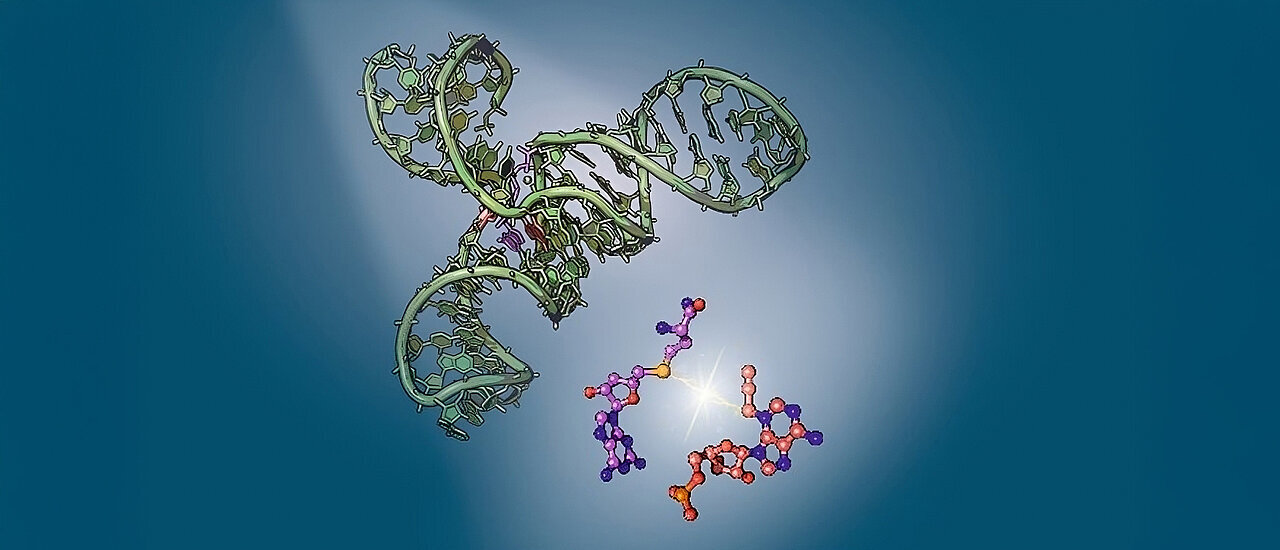
Metal-metal bonding plays a crucial role in the properties and reactivity of molecules composed of metallic atoms. These bonds are especially significant in molecules containing transition metals, such as iridium (Ir), which is known for its unique electronic structure and diverse reactivity. One of the intriguing features of iridium-based molecular systems is the existence of isomers—different spatial arrangements of the same atoms, each with distinct chemical properties. These isomers can have a significant impact on the behavior of the molecules in catalysis, sensing, and other applications. Despite years of research, accurately predicting the proportions of these isomers and their interactions has remained a challenge.
Recent advancements in experimental techniques and theoretical modeling have provided new insights into the behavior of iridium-iridium (Ir-Ir) metal-metal dimer complexes. These complexes are of particular interest due to their potential applications in fields such as chemical sensing and catalysis, where the molecular properties are closely linked to the precise arrangement of atoms and the distances between them. By combining ultrafast experimental measurements with numerical simulations, researchers have made significant strides in understanding how these isomers form, interact, and equilibrate.
Metal-Metal Dimer Complexes and Their Isomers
Iridium metal dimers, specifically those with a d8 square planar metallic center, represent a class of molecules that exhibit unique chemical and physical properties due to the metal-metal bonding interactions. These properties are highly dependent on the spatial arrangement of the iridium atoms, which can adopt different geometries known as isomers. In particular, the Ir-Ir complex can exist in two main configurations, commonly referred to as the “short” and “long” configurations. These terms describe the distance between the iridium atoms in each geometry, with the short configuration having a closer Ir-Ir distance and the long configuration having a more extended structure.
The different spatial arrangements of these dimers have a profound impact on their optical and reactive properties, making them potentially useful in a variety of applications, including as catalysts and sensors. However, to fully harness the potential of these molecules, it is essential to understand and predict the equilibrium between these two configurations, as well as how they influence the molecule’s reactivity. Despite the significance of these isomers, it has been difficult for researchers to precisely predict their proportions in equilibrium or understand the forces that drive their interconversion.
Unveiling the Dynamics of Iridium Isomers: A Combined Approach
The study of these Ir-Ir complexes has taken a significant leap forward with a novel approach that combines experimental techniques with computational simulations. By employing ultrafast X-ray solution scattering, researchers have gained valuable insight into the dynamics of these isomers. The Linac Coherent Light Source (LCLS), a state-of-the-art facility funded by the U.S. Department of Energy’s Office of Science, played a key role in this research. The LCLS produces ultrashort pulses of high-energy X-rays that allow scientists to capture the fast-moving dynamics of molecules in real time, providing the necessary time and spatial resolution to track the re-equilibration of the Ir-Ir isomers.
In this study, the researchers used an innovative technique known as optical hole burning, a process in which one of the isomers—specifically, the long configuration—was selectively depleted from the system. This created an imbalance in the population of the two isomers, prompting the system to seek equilibrium by transitioning from the long isomer back to the short isomer, and vice versa. By tracking this re-equilibration process with ultrafast X-ray solution scattering, the researchers were able to extract important information about the equilibrium proportions of the two isomers and the transition rates between them.
This experimental data provided crucial insights into the dynamics of Ir-Ir complexes, revealing the forces at play in the ultrafast equilibration of metal-metal bonding. In addition to the experimental findings, the researchers used numerical simulations to model the system and compare different theoretical approaches to predicting the equilibrium isomer proportions.
Numerical Simulations and Density Functional Theory (DFT)
At the heart of the computational aspect of this study was the use of density functional theory (DFT), a quantum mechanical modeling approach widely used to study the electronic structure of molecules and materials. DFT is an essential tool for understanding the interactions between atoms in a molecule and predicting the properties of different molecular configurations. However, not all DFT approximations are created equal, and the choice of approximation can significantly affect the accuracy of the predictions.
In this study, the researchers compared several different DFT approximations with the experimental data obtained from the ultrafast X-ray solution scattering. By doing so, they were able to assess which approximations most accurately modeled the dynamics and equilibrium of the Ir-Ir complexes. The findings revealed that certain DFT approximations were better at predicting the observed experimental results, particularly in terms of the proportions of the two isomers present in the ground state.
The ability to accurately predict the equilibrium isomer proportions and transition rates is a significant step forward in the study of metal-metal bonding. By identifying the best DFT approximations for this system, the researchers have established a more reliable method for modeling metal-metal interactions in Ir-Ir and similar complexes. This, in turn, will help guide the design of new molecules for use in various applications, such as catalysis, where precise control over molecular geometry and reactivity is critical.
Implications for Sensing and Catalysis
The ability to predict and control the isomeric forms of Ir-Ir metal dimers has important implications for their potential applications in sensing and catalysis. In these fields, the properties of a molecule are often closely tied to its spatial arrangement and the distances between metal atoms. For example, in catalytic reactions, the distance between metal centers can influence the activation energy and reaction rates, while in sensing applications, the optical properties of the molecule can be tuned by altering its geometry.
Understanding the forces that drive the equilibration of metal-metal dimers and how to predict the equilibrium proportions of isomers will be crucial for optimizing these molecules for specific applications. The research presented here provides a systematic approach to modeling these systems, paving the way for the development of configurable molecules with tailored properties. This could lead to more efficient catalysts and more sensitive sensors, with a wide range of applications in industries such as environmental monitoring, energy production, and chemical manufacturing.
Reference: Natalia E. Powers-Riggs et al, Characterization of Deformational Isomerization Potential and Interconversion Dynamics with Ultrafast X-ray Solution Scattering, Journal of the American Chemical Society (2024). DOI: 10.1021/jacs.4c00817