Every time you swallow a pill—whether it’s for pain, infection, or chronic illness—there’s a molecular gatekeeper working behind the scenes that may determine how well that medicine works. It’s called Cytochrome P450, or CYP, a family of proteins that metabolize over 80% of FDA-approved drugs. Among them, CYP3A4 is the heavy hitter, breaking down some of the most widely used medications. But here’s the catch: if CYP3A4 chews through a drug too fast, it can reduce its effectiveness.
For years, scientists have tried to slow this down by using CYP inhibitors—compounds designed to block CYP3A4’s activity and boost drug efficacy. The problem? These inhibitors often hit the wrong targets too, blocking other CYPs like CYP3A5 and causing unwanted, even dangerous, side effects.
Now, researchers at St. Jude Children’s Research Hospital have made a breakthrough. They’ve engineered drug scaffolds that zero in on CYP3A4 while leaving other CYP proteins largely untouched. Published in Nature Communications, this research marks a major step forward in drug design—one that could enhance medication safety and effectiveness for millions of patients.
The Trouble with CYPs: Why Selectivity Matters
Imagine trying to turn off a single light switch in a room full of switches—and instead you hit half the circuit breaker. That’s essentially what happens when nonselective CYP inhibitors are used.
These inhibitors are meant to slow CYP3A4 so drugs like paclitaxel (used in cancer treatment) or nirmatrelvir (part of the COVID-19 drug Paxlovid) can stay active in the body longer. However, because CYP3A4 and CYP3A5 share similar structures—especially large, flexible binding pockets—many inhibitors unintentionally hit both.
This matters. Blocking CYP3A5 or other CYPs can lead to dangerously high drug levels in the bloodstream, increasing the risk of toxicity, side effects, or drug-drug interactions.
“When a nonselective CYP3A inhibitor is used… the unnecessary inhibition of other CYPs will lead to dangerously elevated plasma levels of drugs metabolized by the unintended CYPs,” explains Dr. Taosheng Chen, senior author and chemical biology expert at St. Jude.
From Thousands to Three: The Quest for Selectivity
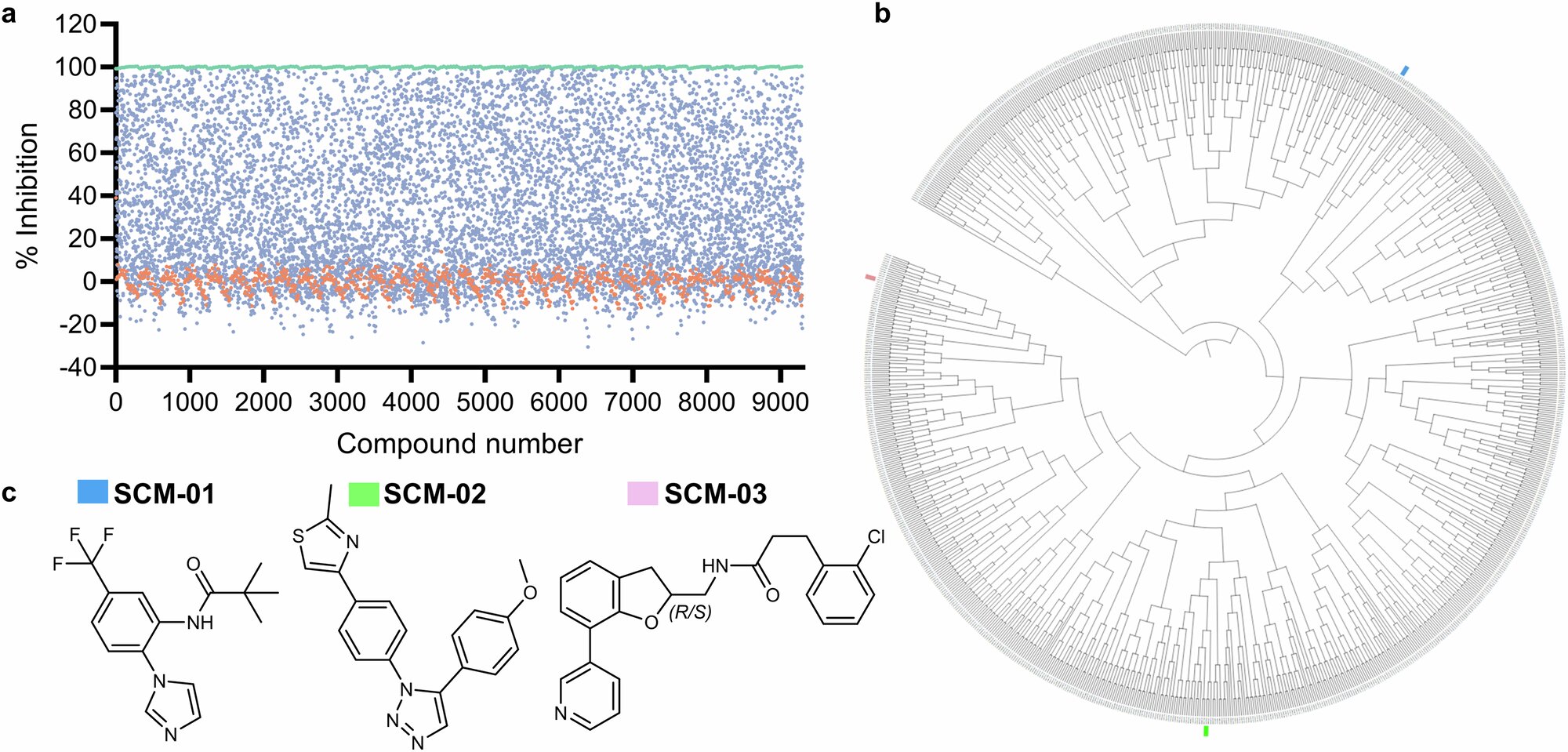
To solve this puzzle, the St. Jude team started with a massive search: a high-throughput screen of 9,299 compounds. From this chemical haystack, they found a needle—or rather, three needles: a trio of inhibitor scaffolds that showed strong, selective inhibition of CYP3A4 with minimal impact on other CYP proteins.
But identifying these molecules was only part of the challenge. Understanding why they worked was the next frontier.
The Structural Secret: A Loop That Makes All the Difference
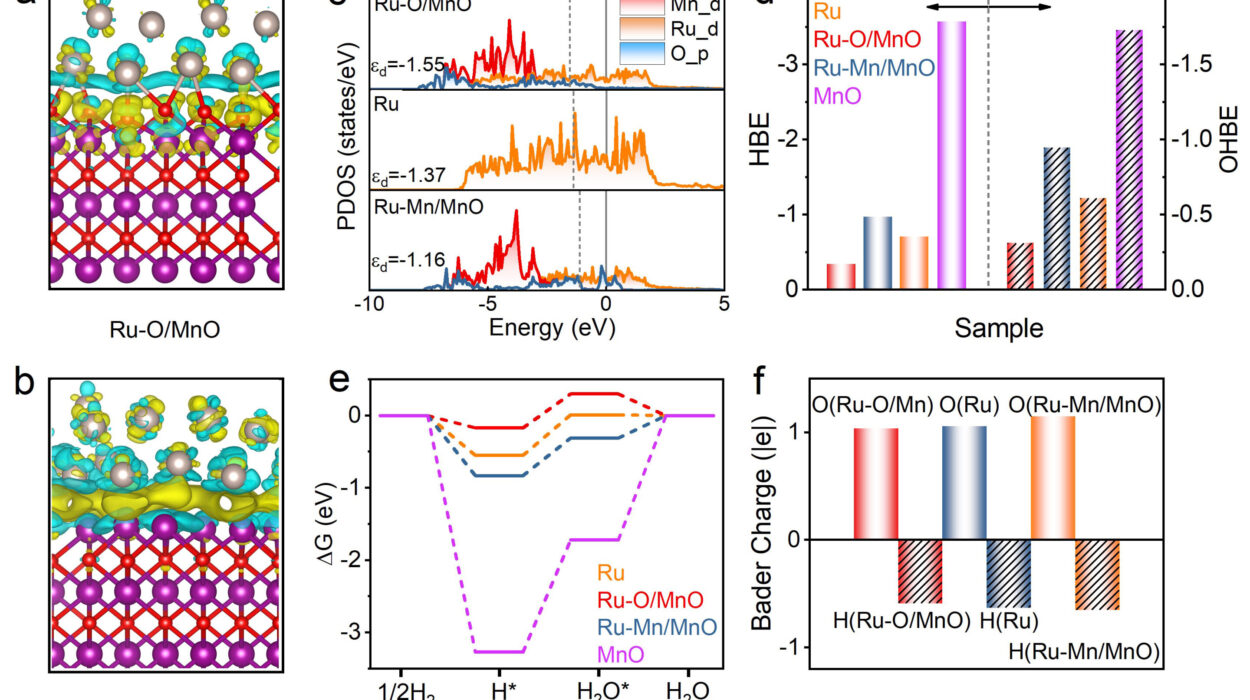
Using X-ray crystallography, the researchers compared the three-dimensional structures of CYP3A4 and CYP3A5. What they discovered was both elegant and game-changing: a loop at the C-terminus of CYP3A5 that acts like a physical shield.
This loop makes the CYP3A5 binding pocket narrower, effectively blocking many of the compounds that would otherwise fit neatly into CYP3A4. That subtle difference allowed the team to rationally tweak their inhibitors for greater precision and fewer off-target effects.
“There is a narrower binding pocket for CYP3A5, which prevents CYP3A4 inhibitors from binding,” said Dr. Chen.
Meet SCM-08: A Blueprint for Better Inhibitors
Among the newly optimized compounds, one stood out: SCM-08. It demonstrated a 46-fold preference for inhibiting CYP3A4 over CYP3A5—an unprecedented level of selectivity. Even more impressive, SCM-08 largely avoided interacting with other CYP enzymes that typically fall victim to broad-spectrum inhibitors.
This means researchers now have a highly selective tool to study and control CYP3A4’s activity in the body—without the collateral damage.
“Our goal is to improve the potency but maintain the selectivity of our CYP3A4-selective inhibitors,” said Chen. “These compounds are the starting points for achieving that, which is now feasible because of the structural basis of selectivity we have uncovered.”
Why This Breakthrough Matters
Selective CYP3A4 inhibitors like SCM-08 could change how clinicians manage drug interactions, especially in cases where co-administered drugs compete for the same metabolic pathways. It could mean:
- More effective treatments by prolonging the activity of fast-metabolized drugs.
- Fewer side effects by sparing other enzymes from unintended inhibition.
- Improved drug design by offering a clearer understanding of CYP selectivity.
This could be especially useful in polypharmacy, where patients—especially the elderly—take multiple drugs that compete or interact. With better control over drug metabolism, doctors could tailor treatments more precisely and safely.
Looking Ahead: From Bench to Bedside
The study’s impact reaches beyond just one protein. By revealing the structural dynamics that govern CYP selectivity, the St. Jude team has provided a roadmap for future drug developers. Their findings could guide the design of selective inhibitors across the broader CYP family, opening new doors for precision medicine.
While SCM-08 and its relatives are still in early stages of development, their potential is enormous. With further refinement and clinical testing, these inhibitors could one day become essential tools in everything from cancer care to infectious disease treatment.
The People Behind the Science
This groundbreaking research was the result of a collaborative effort. Jingheng Wang, Stanley Nithianantham, Sergio Chai, and Young-Hwan Jung led the study as co-first authors, alongside a team of skilled scientists including Lei Yang, Han Wee Ong, Yong Li, Yifan Zhang, and Darcie Miller, all working under the leadership of Dr. Taosheng Chen at St. Jude’s Department of Chemical Biology & Therapeutics.
Their work reflects not just a major scientific advance, but a powerful reminder of the role structural biology can play in solving real-world medical problems.
Reference: Jingheng Wang et al, Decoding the selective chemical modulation of CYP3A4, Nature Communications (2025). DOI: 10.1038/s41467-025-58749-8